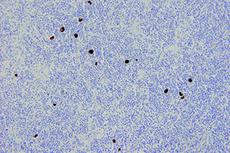
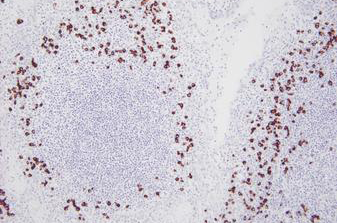
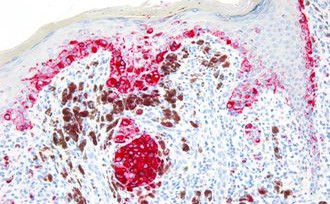
Advancing Cancer Diagnostics, Improving Lives
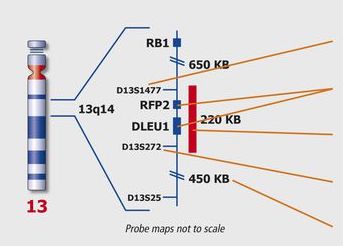
Kreatech FISH Probes
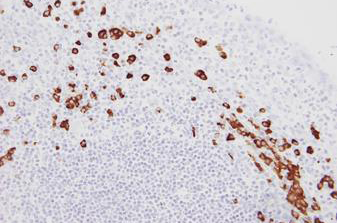
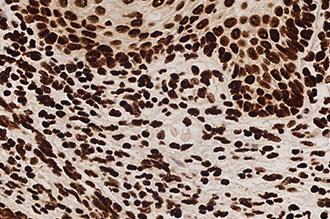
Kreatech FISH probes are the latest in advancements in FISH probes. Developed with the use of REPEAT-FREE technology, Kreatech FISH probes eliminate the use of Cot-1 or blocking DNA, providing a clearer background and a brighter signal.
Novocastra FISH Probes
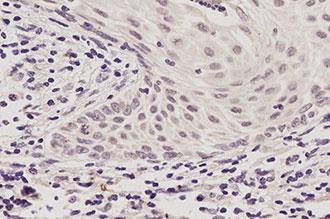
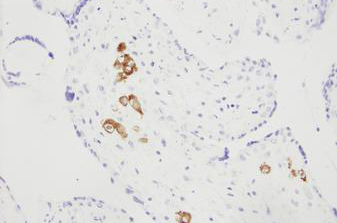
Reliably detect RNA sequences with Novocastra ISH probes, detection systems, and ancillary reagents.